Pharma Manufacturing

Agentic AI in pharma: Separating the hype from reality. Learn how to scale autonomous R&D workflows in drug development.
Simantini Singh Deo | Jul 04, 2026
How AI Is Making Personalized Medicine Scalable?
Simantini Singh Deo | Jul 04, 2026

Difference Between Hard and Soft Gelatin Capsules
Mrudula Kulkarni | Jul 04, 2026
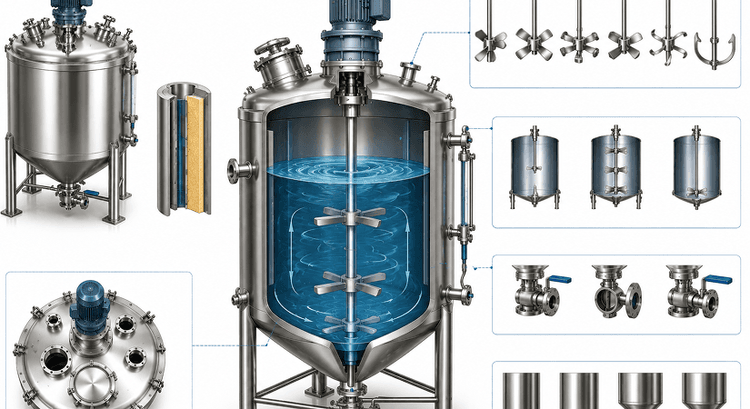
A Guide on Industrial Mixing Tank Stirrer Specification
Mrudula Kulkarni | Jul 02, 2026
Other trending pieces on pharma manufacturing you may like to read
Agentic AI In Drug Development: Hype Or Reality?
Agentic AI in pharma: Separating the hype from reality. Learn how to scale autonomous R&D workflows in drug development.
by Simantini Singh Deo

How AI Is Making Personalized Medicine Scalable?
AI is solving the personalized medicine bottleneck. Learn how ML virtual screening scales precision oncology and targets the undruggable.
by Simantini Singh Deo
Difference Between Hard and Soft Gelatin Capsules
Hard gelatin capsules hold powders. Softgels hold oils. Here's the full comparison, structure, fill, manufacturing, cost, and bioavailability.
by Mrudula Kulkarni

A Guide on Industrial Mixing Tank Stirrer Specification
Impeller selection, P/V, surface finish, ATEX, CIP/SIP — the complete industrial mixing tank specifications with stirrer systems for pharma engineers.
by Mrudula Kulkarni

The Great Pharma Manufacturing Shift: Why Localisation Matters More Than Ever
Pharma supply chain resilience now demands localisation, why regional manufacturing is shifting from contingency to core strategy.
by Vaibhavi M.
